Diseases
Click here to view more Diseases
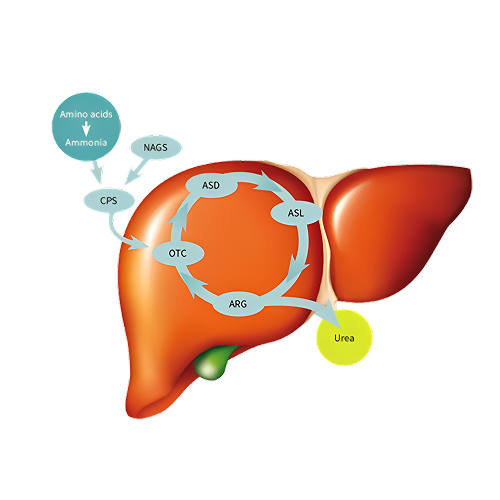
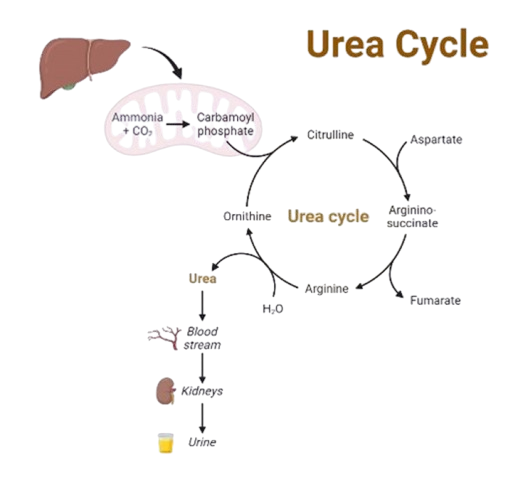
Urea cycle disorders (UCDs) are inborn errors of metabolism resulting from defects in one of the enzymes or transporter molecules involved in the hepatic removal of ammonia from the bloodstream. Removal of ammonia from the bloodstream normally occurs via its conversion to urea, which is then excreted by the kidneys. Consequently, urea cycle disorders lead to an accumulation of ammonia. Ammonia is extremely toxic, particularly to the central nervous system. Newborns with severe mutations in any one of the first four enzymes of the urea cycle can become catastrophically ill within 36 to 48 hours of birth despite appearing normal at birth.
Urea cycle defects (UCDs) are a group of rare inborn errors of metabolism
with a
cumulative
incidence of 1:35,000–
1:69,000. Overall, the incidence of UCDs has recently been determined to be
1 in
35,000 births.
Urea cycle disorder and
hyperammonemia have been cause estimated to be 1 per 53,717 (i.e.)
approximately 1.9
per 100,000
live births in India.
In India, all types of UCDs the incidence of the disorder is approximately 1
in
10,000 births,
also indicating a
noticeable surge in case identification and diagnosis in the past few years.
Most
state screening
laboratories currently
detect only 2 of all the conditions that cause UCDs. About two-thirds of all
USDs
are due to
mutations in ornithine
transcarbamylase (OTC), one-fifth due to argininosuccinate synthase 1(ASS1),
and
one-tenth due to
argininosuccinate
lyase (ASL)
Is the sole source of endogenous production of arginine, ornithine, and citrulline; it is the principal mechanism for the clearance of waste nitrogen resulting from protein turnover. Urea cycle disorders (UCDs) result from inherited deficiencies in any one of the six enzymes or two transporters of the urea cycle pathway (CPS1, OTC, ASS1, ASL, ARG1, NAGS, ORNT1, or citrin).
N-acetyl glutamate synthetase (NAGS)
Severe deficiency or total absence of activity of any of the first four enzymes in the pathway (CPS1, OTC, ASS1, and ASL) or the cofactor producer (NAGS) results in the accumulation of ammonia and other precursor metabolites during the first few days of life. Because no effective secondary clearance system for ammonia exists, complete disruption of this pathway results in the rapid accumulation of ammonia and development of related symptoms.
There has been considerable progress in understanding the pathophysiology of UCDs, which may ultimately promote better medical interventions. Hyperammonemia is a key etiological factor in UCDs and plays a key role in CNS toxicity. Although complex, some important concepts are emerging. The toxic effects of ammonia on the CNS are more severe in the developing brain than in the adult brain.
Whether the toxic effects of hyperammonemia are reversible or not depends upon the exposure time, the dose of hyperammonemia, and the stage of neurological development. In the neonatal brain, hyperammonemia results in edema caused by swelling of astrocytes. The subsequent frequency and degree of swelling are key factors in determining the severity of CNS neurological disorders such as seizures, coma, and cognitive/motor deficits.
While OTC deficiency (OTCD) is inherited in an X-linked manner, all other UCDs follow an autosomal recessive pattern. Moreover, there is evidence that even asymptomatic female carriers of OTCD can develop cognitive defects as a result of episodic hyperammonemia.
Presentation. Individuals with complete defects normally present in the newborn period, when the immaturity of the neonatal liver accentuates defects in the urea cycle enzymes.
The typical initial symptoms of a child with hyperammonemia are nonspecific Failure to feed
Symptoms progress from somnolence to lethargy and coma.
| Indication | Dose | Contraindications | Adverse Effects |
|---|---|---|---|
| Sodium phenylbutyrate powder is indicated as adjunctive therapy in the chronic management of patients with urea cycle disorders. It is indicated in all patients with neonatal-onset deficiency (complete enzymatic deficiency, presenting within the first 28 days of life). It is also indicated in patients with late-onset disease (partial enzymatic deficiency, presenting after the first month of life) who have a history of hyperammonemic encephalopathy. | The usual total daily dose of sodium phenylbutyrate powder for patients with urea cycle disorders is 450 to 600 mg/kg/day in patients weighing less than 20 kg, or 9.9 to 13 g/m2/day in larger patients. The powder is to be taken in equally divided amounts with each meal or feeding (i.e., three to six times per day). | Sodium phenylbutyrate, USP should not be used to manage acute hyperammonemia, which is a medical emergency. | In female patients, the most common clinical adverse event reported was
amenorrhea/menstrual dysfunction (irregular
menstrual cycles), which occurred in 23% of the menstruating patients. Decreased appetite occurred in 4% of all patients. Body odor (probably caused by the metabolite, phenylacetate) and bad taste or taste aversion were each reported in 3% of patients. Other adverse events reported in 2% or fewer patients were: Gastrointestinal, Hematologic, Cardiovascular, Renal and Psychiatric: depression etc. |
Gene therapy for UCDs is rapidly expanding. Due to the severity of the clinical phenotype, favourable risk benefit profile for experimental intervention and relatively common presentation in the field of inherited metabolic diseases, most pioneering liver‐targeting technologies will explore applications as proof of concept in UCDs. New innovations in urea cycle disorders (UCDs) are focused on gene therapy and gene editing, as well as exploring RNA therapies to improve treatment outcomes. Gene therapy, particularly when combined with gene editing, shows promise in addressing the core issue of UCDs: enzyme deficiencies that lead to ammonia buildup. Additionally, N-carbamylglutamate, an oral medication, has demonstrated the ability to correct biochemical defects in certain UCDs.
Urea cycle defects (UCDs) are severe inherited metabolic diseases with high unmet needs which present a permanent risk of hyperammonaemic decompensation and subsequent acute death or neurological sequelae, when treated with conventional dietetic and medical therapies. Liver transplantation is currently the only curative option, but has the potential to be supplanted by highly effective gene therapy interventions without the attendant need for life-long immunosuppression or limitations imposed by donor liver supply. Over the last three decades, pioneering genetic technologies have been explored to circumvent the consequences of UCDs, improve quality of life and long-term outcomes.
Future research directions for urea cycle disorders (UCDs) focus on developing more effective treatments beyond current options like dietary management and liver transplantation. This includes exploring gene therapy, improved diagnostic methods, and better understanding of the impact of hyperammonemia on brain function.

Disclaimer
 Healthcare
Professional
Healthcare
Professional